жң¬её–жңҖеҗҺз”ұ еһ…з”°жҘјдёҖ дәҺ 2015-6-24 01:17 зј–иҫ‘ : b$ ~3 K$ V& b2 l
9 }! S& L L" S
еҗҙдёҖйҫҷпјҡиӮәзҷҢз»јеҗҲжІ»з–— 2014-03-30 20:42 жқҘжәҗпјҡдёҒйҰҷеӣӯ 2 h3 f3 K- h8 m! Z1 ?( w
5 N7 r7 q4 A- ] зј–иҖ…жҢүпјҡиӮәзҷҢжҳҜдёҘйҮҚеҚұе®іжҲ‘еӣҪдәәж°‘иә«дҪ“еҒҘеә·зҡ„дё»иҰҒзҷҢз§Қд№ӢдёҖпјҢеҸ‘з—…зҺҮеңЁжҲ‘еӣҪе‘ҲдёҠеҚҮи¶ӢеҠҝпјҢ
- d2 g& F) n4 I7 `5 @е…¶з»јеҗҲжІ»з–—зҗҶеҝөйҖҗжёҗдёәдёҙеәҠе№ҝжіӣйҮҮзәігҖӮдёәдәҶи§ЈиӮәзҷҢз»јеҗҲжІ»з–—зҡ„жңҖж–°иҝӣеұ•пјҢеӨҡеӯҰ科еҗҲдҪңзҡ„йҮҚиҰҒжҖ§пјҢ/ e5 ]1 r, |( u8 q3 l7 l) j, H* P
д»ҘеҸҠеӨ–科治疗зҡ„еҸ‘еұ•ж–№еҗ‘пјҢдёҒйҰҷеӣӯи®°иҖ…еңЁйҰ–еұҠиғёйғЁиӮҝзҳӨиҘҝеӯҗи®әеқӣдёҠеҜ№еҗҙдёҖйҫҷж•ҷжҺҲиҝӣиЎҢдәҶдё“и®ҝгҖӮ {; }+ h9 [; k* X$ F) b8 X9 B. u
9 h8 p# J" A; w& P: h8 K/ V
7 d/ }% `9 h# v8 Y$ w. A
еҗҙдёҖйҫҷж•ҷжҺҲпјҢе№ҝдёңзңҒдәәж°‘еҢ»йҷўеүҜйҷўй•ҝгҖҒе№ҝдёңзңҒиӮәзҷҢз ”з©¶жүҖжүҖй•ҝгҖҒе№ҝдёңзңҒиӮҝзҳӨйҳІжІ»дёӯеҝғдё»д»»гҖҒдёӯеұұеӨ§еӯҰиӮҝзҳӨеӯҰж•ҷжҺҲ* V' D3 V% [6 ~4 b
' M. ~5 x* d- e# H) e# bдёҒйҰҷеӣӯпјҡеҗҙж•ҷжҺҲжӮЁеҘҪпјҢйқһеёёж„ҹи°ўжӮЁжҺҘеҸ—дёҒйҰҷеӣӯзҡ„дё“и®ҝгҖӮиӮәзҷҢз»јеҗҲжІ»з–—жҳҜиҝҷж¬ЎеӨ§дјҡзҡ„дә®зӮ№д№ӢдёҖ, ?1 @+ f! r+ D4 ?/ d+ p2 V
жӮЁиғҪи°Ҳи°ҲиӮәзҷҢз»јеҗҲжІ»з–—зҡ„жңҖж–°иҝӣеұ•еҗ—пјҹжӮЁдёӘдәәеҰӮдҪ•зҗҶи§ЈеӨҡеӯҰ科еҗҲдҪңеңЁиӮәзҷҢжІ»з–—дёӯзҡ„йҮҚиҰҒжҖ§пјҹ
, F" K# n) w: O& B1 s; b x6 j. l6 u! u( W4 a
еҗҙдёҖйҫҷж•ҷжҺҲпјҡиӮәзҷҢзҡ„з»јеҗҲжІ»з–—дёҚеӨ–д№ҺеҰӮдҪ•еҗҲзҗҶең°жҠҠзҺ°жңүзҡ„жІ»з–—жүӢж®өз»јеҗҲеҲ°дёҖиө·гҖӮ* g, z# y% [2 h- a I; E/ C% a
еҫҲеӨҡдәәжҠҠз»јеҗҲжІ»з–—з®ҖеҚ•ең°зҗҶи§ЈдёәжүӢжңҜжІ»з–—гҖҒеҢ–еӯҰжІ»з–—гҖҒж”ҫе°„жІ»з–—зҡ„еҸ еҠ гҖӮ
9 A, I- Y) M C/ l7 d# Kе®һйҷ…дёҠпјҢзҺ°д»Јз»јеҗҲжІ»з–—зҡ„зҗҶеҝөејәи°ғеҜ№еҗ„з§ҚжІ»з–—ж–№жі•зҡ„й•ҝеӨ„дёҺзҹӯеӨ„йғҪиҰҒдәҶи§ЈпјҢ
) ?' A) ]8 f* {- x然еҗҺж №жҚ®иӮҝзҳӨзү№еҲ«жҳҜиӮәзҷҢзҡ„з”ҹзү©еӯҰиЎҢдёәпјҢжҠҠиҝҷдәӣжүӢж®өжңүжңәең°з»“еҗҲиө·жқҘгҖӮ. [- |% z# N, b3 i: @! l
' N" U& @2 @9 N9 _
жҜ”еҰӮиҜҙд»ҠеӨ©жҸҗеҮәжқҘзҡ„ж··еҗҲз–—ж•Ҳй—®йўҳпјҢж„Ҹе‘ізқҖзҷҢз—Үзү№еҲ«жҳҜиӮәзҷҢзҡ„з»јеҗҲжІ»з–—еңЁжңӘжқҘеҸҜд»Ҙй—ҜеҮәдёҖжқЎеҙӯж–°зҡ„йҒ“и·ҜгҖӮ/ A) P6 ^" o1 W; {- t% D
иҝҮеҺ»и®ӨдёәеұҖйғЁжІ»з–—еҜ№жҷҡжңҹиӮәзҷҢеҮ д№ҺжҳҜжІЎжңүдҪҚзҪ®зҡ„пјҢз—…дәәдёҖж—ҰеҸ‘з”ҹе…Ёиә«иҪ¬з§»е°ұжІЎжңүеұҖйғЁжІ»з–—зҡ„ж„Ҹд№үдәҶгҖӮ3 }" h# w* h" S
дҪҶжҳҜпјҢзҺ°еңЁжҲ‘们и®ӨиҜҶеҲ°иӮҝзҳӨзҡ„з”ҹзү©еӯҰиЎҢдёәжҳҜдёҖз§Қж··еҗҲз–—ж•ҲпјҢ6 K' T* M$ o4 f9 u x1 f
еңЁе…Ёиә«з–ҫз—…жҺ§еҲ¶иүҜеҘҪзҡ„жғ…еҶөдёӢеҜ№еұҖйғЁиҝӣиЎҢеӨ„зҗҶдјҡеҸ–еҫ—жӣҙеҘҪзҡ„ж•ҲжһңгҖӮ
& w/ p0 e1 `& U+ T9 h, j dиҝҷе°ұжҳҜеҜ№з”ҹзү©еӯҰжңүдәҶж·ұеҲ»и®ӨиҜҶд№ӢеҗҺжҸҗеҮәзҡ„з»јеҗҲжІ»з–—зҗҶеҝөпјҢиҝҷе’ҢиҝҮеҺ»жҳҜдёҚдёҖж ·зҡ„гҖӮ
& R7 U9 g% S2 }, J1 w% k
2 M' M8 b; d, w+ M0 Sиҝҷз§Қжғ…еҶөзҡ„еҮәзҺ°иҝҷжңүиө–дәҺеҗ„дёӘеӯҰ科жҠҖжңҜжң¬иә«зҡ„иҝӣжӯҘгҖӮ; v% J. Z/ a; n
зҺ°еңЁйҷӨдәҶеҢ–з–—иҚҜзү©пјҢд№ҹжңүйқ¶еҗ‘иҚҜзү©пјҢз–—ж•Ҳеҫ—еҲ°жһҒеӨ§жҸҗй«ҳгҖӮ: q% H! I G# P, {% L3 a4 D) {
зҺ°еңЁзҡ„иғёи…”й•ңжүӢжңҜпјҢе®һзҺ°дәҶеҫ®еҲӣпјҢдҪҝеұҖйғЁжІ»з–—еҜ№жңәдҪ“зҡ„жҚҹдјӨйҷҚеҲ°дәҶжңҖдҪҺгҖӮ( d& A4 T F3 k& p
д»ҠеӨ©зҡ„ж”ҫе°„жІ»з–—пјҢзІҫеҮҶеәҰеӨ§еӨ§жҸҗй«ҳпјҢеҜ№жӯЈеёёз»„з»Үзҡ„жҚҹдјӨеӨ§еӨ§еҮҸе°ҸгҖӮ7 `3 H/ Y3 E3 }" O: E
жӯЈжҳҜиҝҷдәӣж–°жҠҖжңҜзҡ„еҮәзҺ°пјҢдҪҝеҫ—еұҖйғЁжІ»з–—еҸ‘жҢҘжӣҙеӨ§зҡ„дҪңз”ЁгҖӮ
k# u4 z' f! E* E: j* ]3 G/ r- l+ f+ k9 S* q: d4 S
иӮәзҷҢзҡ„з»јеҗҲжІ»з–—е·Із»ҸеҸ‘еұ•еҲ°дәҶдёҖдёӘж–°зҡ„йҳ¶ж®өпјҢиҝҷзҰ»дёҚејҖеҜ№иӮҝзҳӨз”ҹзү©еӯҰзҡ„и®ӨиҜҶпјҢ9 k" h( y, Z& w3 L
зҰ»дёҚејҖеҗ„з§ҚжІ»з–—жүӢж®өзҡ„зІҫз»ҶеҢ–гҖӮ
_# W' z, D! ]( J- o5 ~7 P
$ R; M) H8 g! y7 N6 _2 p+ B6 h/ iеңЁиӮәзҷҢжІ»з–—дёӯдёҖе®ҡйңҖиҰҒеӨҡеӯҰ科еҗҲдҪңгҖӮ
: n$ [0 f- |! c! [д»Ҙж—©жңҹиӮәзҷҢдёәдҫӢпјҢиҝҮеҺ»еҚ•зәҜеҒҡдёӘжүӢжңҜе°ұеҸҜд»ҘдәҶпјҢд»ҠеӨ©дҫқ然йҖүжӢ©еҚ•зәҜзҡ„жүӢжңҜжІ»з–—пјҢ
' [1 J: L: z' X* H- N rдҪҶжҳҜжҲ‘们已з»Ҹжё…жҘҡең°и®ӨиҜҶеҲ°иҝҷдёӘж—¶еҖҷеҶҚеҠ дёҖдәӣжІ»з–—еҜ№з—…дәәжІЎжңүеҘҪеӨ„пјҢеҸӘжңүеқҸеӨ„гҖӮ1 K) E! T1 a A0 O( p3 L! x
иҝҮеҺ»дёҚеҠ жІ»з–—е’Ңд»ҠеӨ©зҹҘйҒ“дёәд»Җд№ҲдёҚеҠ жІ»з–—е·Із»ҸжңүдәҶеҫҲеӨ§жҸҗй«ҳгҖӮ
: q$ Q9 C4 V9 f* Y! y: L2 E# o- A' iжүҖд»ҘпјҢз»јеҗҲжІ»з–—еңЁзҗҶи®әдёҠжңүдәҶиҙЁзҡ„еҸҳеҢ–гҖӮ, K3 v4 H3 }, e; ~5 v: z& s4 r
" r; F1 h) d: J" K) TдёҒйҰҷеӣӯпјҡжӮЁеҲҡеҲҡеңЁеӨ§дјҡдёҠи®ІеҲ°иӮәзҷҢејӮиҙЁжҖ§е’ҢжІ»з–—зӯ–з•ҘпјҢ0 d- H% |6 h' w2 k& J, g+ J, Z
зӣ®еүҚеңЁиӮәзҷҢзҡ„з»јеҗҲжІ»з–—жңүжІЎжңүе…ұиҜҶжҲ–жҳҜ规иҢғеҢ–зҡ„иҜҠз–—жҖқз»ҙпјҹ
2 A! D; [8 m1 yйӮЈеұҖйғЁжҷҡжңҹйқһе°Ҹз»ҶиғһиӮәзҷҢз»јеҗҲжІ»з–—зӯ–з•ҘеҸҲжңүе“ӘдәӣиҰҒзӮ№пјҹ
( r9 ^' e+ B3 f, u2 G. j O" T( K! G6 E 8 n! }1 @+ L: h9 a9 F+ d
еҗҙдёҖйҫҷж•ҷжҺҲпјҡиӮәзҷҢзҡ„з»јеҗҲжІ»з–—зӯ–з•ҘдёҖзӣҙеңЁдёҚж–ӯеҸ‘еұ•гҖӮ
3 K o, k, h5 eдёҫдёӘз®ҖеҚ•зҡ„дҫӢеӯҗпјҢиӮәзҷҢжҷҡжңҹзҡ„з—…дәәдјҡеҮәзҺ°иҖҗиҚҜжҖ§гҖӮд»ҠеӨ©жҲ‘们зҹҘйҒ“дәҶиҖҗиҚҜзҡ„жңәеҲ¶йқһеёёеӨҚжқӮпјҢе®ғжңүдёүз§ҚеӨ„зҗҶж–№жі•гҖӮ
" \& U+ e! `! J, `$ ^& j, B, I第дёҖпјҢиҖҗиҚҜиЎЁзҺ°дёәжҹҗдәӣеҹәеӣ дә§з”ҹдәҶзӘҒеҸҳпјҢиҝҷз§ҚзӘҒеҸҳиҰҒй’ҲеҜ№жңәеҲ¶иҝӣиЎҢеӨ„зҗҶпјҢиҝҷжҳҜе…ұиҜҶгҖӮ( }' q) x6 i# z/ U
第дәҢпјҢзӣ®еүҚдёәжӯўиҝҳжүҫдёҚеҲ°иҖҗиҚҜзҡ„жңәеҲ¶пјҢдҪҶжҳҜеҸҜд»Ҙд»ҺдёҙеәҠзҺ°иұЎиҝӣиЎҢеҲҶзұ»пјҢж №жҚ®еҲҶзұ»иҝӣиЎҢдёҚеҗҢзҡ„еӨ„зҗҶгҖӮ! q. Q9 ~/ m, M; l; ^2 r1 y, V
第дёүпјҢдёҖж—Ұиҝӣеұ•е°ұжҚўжҲҗеҸҰеӨ–дёҖз§ҚиҚҜпјҢиҝҷд№ҹжҳҜеӨ§е®¶зӣ®еүҚеҪўжҲҗзҡ„е…ұиҜҶгҖӮ
) O- c* C' h3 F: E, Z6 t1 ^# l/ gжҜҸдёҖж¬Ўе…ұиҜҶйғҪжҳҜеҜ№дәӢзү©зҡ„и®ӨиҜҶжӣҙж·ұеҲ»д№ӢеҗҺжүҚеҪўжҲҗзҡ„гҖӮ/ l2 z7 | E0 @: J$ Z$ {5 l, M$ }
6 k7 Z1 h; o, T6 K# ]% \. i- \
жҳҺе№ҙдёүжңҲд»ҪеңЁе№ҝе·һдёҫиЎҢ第еҚҒеұҠдёӯеӣҪиӮәзҷҢй«ҳеі°и®әеқӣпјҢйҮҚзӮ№е°ұжҳҜеҪўжҲҗеҜ№иҖҗиҚҜзҡ„еӨ„зҗҶе…ұиҜҶгҖӮ
. _6 l6 j- z0 l. ]
& w) c! c. O5 D: p- N$ @5 I. D* qжҲ‘и®ӨдёәејӮиҙЁжҖ§еӯҳеңЁдәҺиӮәзҷҢзҡ„еҗ„дёӘйҳ¶ж®өгҖӮеңЁиӮәзҷҢзҡ„ж—©жңҹгҖҒеұҖйғЁжҷҡжңҹгҖҒжҷҡжңҹпјҢйғҪиҰҒиҖғиҷ‘еҲ°ејӮиҙЁжҖ§зҡ„й—®йўҳгҖӮ
, ~8 W6 W4 L2 G$ `' B( a8 G5 RеҪ“жҲ‘们用дёҖз§Қж–№жі•и§ЈеҶідёҚдәҶй—®йўҳзҡ„ж—¶еҖҷеҸҜиғҪиҰҒиҖғиҷ‘еҲ°ејӮиҙЁжҖ§зҡ„й—®йўҳгҖӮ3 R/ [+ _) |. j% |4 z
иҝҮеҺ»и®ӨдёәпјҢдёҖз§ҚжІ»з–—жүӢж®өжІЎжңүж•Ҳжһңе°ұжҳҜиҝҷз§ҚжІ»з–—жүӢж®өзҡ„еӨұиҙҘпјҢ+ E: H a8 I7 P/ w* E* W
е…¶е®һиҝҷз§ҚжІ»з–—жүӢж®өеҜ№дёҖйғЁеҲҶдәәжҳҜжҲҗеҠҹзҡ„пјҢиҖҢеҜ№дёҖйғЁеҲҶдәәжҳҜдёҚжҲҗеҠҹзҡ„пјҢиҝҷе°ұжҳҜејӮиҙЁжҖ§зҡ„й—®йўҳгҖӮ
/ t8 C% X" g/ E e; v4 uжүҖд»ҘиҜҙд»ҠеӨ©жҸҗеҮәејӮиҙЁжҖ§зҡ„й—®йўҳпјҢиҰҒи®ӨиҜҶеҲ°д»ҺиӮәзҷҢж—©жңҹеҲ°еұҖйғЁжҷҡжңҹеҶҚеҲ°жҷҡжңҹпјҢиӮҝзҳӨжғ…еҶөдјҡдә§з”ҹйқһеёёеӨ§зҡ„еҸҳеҢ–гҖӮ' _0 @. ]; ~) G& @8 R
еҪ“жҲ‘们碰еҲ°иҝҷдәӣй—®йўҳж—¶пјҢеҝ…йЎ»иҰҒжғіеҲ°иҝҷжҳҜејӮиҙЁжҖ§зҡ„й—®йўҳгҖӮ2 W5 W1 P* I# f4 `6 d D, @
3 G* V3 B# ?4 _
дёҒйҰҷеӣӯпјҡиғёйғЁиӮҝзҳӨзҡ„иҜҠж–ӯгҖҒеӨ–科жүӢжңҜеҸҠз»јеҗҲжІ»з–—зҡ„еҸ‘еұ•еҰӮзҒ«еҰӮиҚјпјҢ( O/ Z+ f! w( n6 p
зӣ®еүҚеӨ–科治疗еҸ‘еұ•зҡ„зү№зӮ№жҳҜжҖҺж ·зҡ„пјҹ( ^2 Z- |0 S: f1 I& z
жӮЁи®ӨдёәжҳҜеҗҰеҜ№дј з»ҹзҡ„еӨ–科жүӢжңҜжЁЎејҸжҸҗеҮәдәҶжӣҙй«ҳзҡ„иҰҒжұӮпјҹ# |( j1 j% ]$ u& j& N d+ C
& r! w# d1 `; @" r+ ^
* S8 \( b" d* x& RеҗҙдёҖйҫҷж•ҷжҺҲпјҡжҲ‘и§үеҫ—еӨ–科治疗зҡ„еҸ‘еұ•дё»иҰҒжңүдёӨдёӘзү№зӮ№гҖӮ
1 x0 q6 Q) _3 |' j第дёҖпјҢеӨ–科жҠҖжңҜи¶ҠжқҘи¶ҠзІҫз»ҶеҢ–пјҢиҝҷжңүиө–дәҺеӨ–科жҠҖжңҜжң¬иә«зҡ„еҸ‘еұ•гҖӮ0 F+ |9 V1 O) D- k/ x' {; H; V
жҜ”еҰӮд»ҺиҝҮеҺ»зҡ„ејҖиғёжүӢжңҜеҲ°зҺ°еңЁзҡ„еҫ®еҲӣжүӢжңҜпјҢеӨ–科жүӢжңҜеҫ®еҲӣеҢ–пјҢиҝҷе°ұжҳҜдёҖдёӘеҸ‘еұ•зҡ„иҝҮзЁӢгҖӮ
- t/ f% R2 m1 F/ O; g第дәҢпјҢжҲ‘们жӣҙеҠ жё…жҘҡең°зҹҘйҒ“жүӢжңҜеңЁжІ»з–—жҒ¶жҖ§иӮҝзҳӨзҡ„жҜҸдёӘйҳ¶ж®өдёӯжү®жј”зҡ„и§’иүІгҖӮ
4 D) u) s; K# k% G# H5 G/ ~ жҜ”еҰӮиҜҙеңЁж—©жңҹиӮәзҷҢпјҢжүӢжңҜиө·дё»иҰҒзҡ„дҪңз”Ёпјӣ
$ Y+ E) E) t: E3 \ еҲ°дәҶеұҖйғЁжҷҡжңҹпјҢеҸҜиғҪеҸӘиө·йғЁеҲҶзҡ„дҪңз”Ёпјӣ
! K) \* ~% Q( O! ~ u: A( L еҲ°дәҶжҷҡжңҹпјҢеҸӘиө·еұҖйҷҗзҡ„дҪңз”ЁпјҢиҝҷжҳҜеҜ№жүӢжңҜжІ»з–—еҸҠе…¶еҸ‘еұ•зҡ„и®ӨиҜҶгҖӮ6 L/ P4 ~. J5 G `( }
5 k: q' S+ \- u+ }& _/ H! L% y
жүҖд»ҘеӨ–科治疗еҸ‘еұ•зҡ„зү№зӮ№дҪ“зҺ°еңЁдёӨеӨ§ж–№йқўпјҢ
?& d3 j& z# W: u" BдёҖж–№йқўжҳҜеҜ№жҠҖжңҜзҡ„зІҫзӣҠжұӮзІҫпјҢдёҚж–ӯзІҫз»ҶеҢ–пјҢ
" S+ H' j: h) ]/ d7 b5 Z+ VеҸҰдёҖж–№йқўжҳҜд»ҺеӨ§дҪ“жқҘзңӢжҲ‘们зҹҘйҒ“е®ғжҜҸдёӘйҳ¶ж®өжүҖиө·зҡ„дҪңз”Ёе’ҢдҪҚзҪ®гҖӮ! f, D( f F, R
0 O. e$ N- ]/ P8 b
иҝҷдәӣеҸ‘еұ•д№ҹж„Ҹе‘ізқҖеҜ№еӨ–科жүӢжңҜзҡ„иҰҒжұӮжҸҗй«ҳдәҶпјҢжҲ‘们еҝ…йЎ»дёҚж–ӯең°еӯҰд№ ж–°зҡ„жҠҖжңҜпјҢиҝҷд№ҹжҳҜеҫ®еҲӣеӨ–科зҡ„еӯҰд№ еҰӮзҒ«еҰӮиҚјзҡ„еҺҹеӣ гҖӮ7 r" t4 u8 U* M+ n
еҗҢж—¶пјҢеҝ…йЎ»еӯҰд№ иӮҝзҳӨзҡ„з”ҹзү©еӯҰзү№жҖ§пјҢж №жҚ®зү№жҖ§жқҘзңӢзңӢжҲ‘们ж•ҙдёӘеӨ–科зҡ„еҸ‘еұ•гҖӮ
. Q. ^, a# i# f9 H; c% r
& f- [0 D9 y GдёҒйҰҷеӣӯпјҡжӮЁи®ӨдёәжңӘжқҘзҡ„еӨ–科еҢ»з”ҹйңҖиҰҒе…·еӨҮе“Әдәӣдё“дёҡзҙ иҙЁпјҹ
7 h1 ?, h2 Q: C+ Z! m0 H жҖҺж ·жүҚиғҪеҹ№е…»еҮәдёҖеҗҚдјҳз§Җзҡ„еӨ–科еҢ»з”ҹпјҹ! t2 @! e# H; U; C9 R& }
& [; T$ i4 F* u# r8 Z6 z. R- t
' ~) G1 w- K4 l0 O) OеҗҙдёҖйҫҷж•ҷжҺҲпјҡжҲ‘з»ҸеёёиҜҙиҝҷж ·дёҖж®өиҜқпјҢдҪңдёәдёҖеҗҚд»ҺдәӢеӨ–科专дёҡзҡ„еҢ»з”ҹпјҢ
% _# N4 a4 G$ a( i! |* L- h+ BеҰӮжһңиҝҷдёӘжүӢжңҜеҲ«дәәдјҡеҒҡдҪ д№ҹдјҡеҒҡпјҢйӮЈдҪ е°ұжҳҜдёҖдёӘеӨ–科жүӢжңҜеҢ гҖӮ$ b, t" P8 M$ d& b2 I/ s' Z5 ~
: I6 C) L/ ]. ~
еҰӮжһңиҝҷдёӘжүӢжңҜеҲ«дәәдёҚдјҡеҒҡиҖҢдҪ дјҡеҒҡпјҢдҪ зҡ„ж°ҙе№іжҜ”еүҚиҖ…иҰҒй«ҳдёҖзә§гҖӮ
! o9 A8 r$ Z& G0 f# k+ R) i2 _3 P9 m5 H4 [& A7 G% |$ O: R" q& L2 \
еҰӮжһңиҝҷдёӘжүӢжңҜдҪ дјҡеҒҡпјҢеҚҙзҹҘйҒ“дёҚеҸҜд»ҘеҒҡпјҢйӮЈд№ҲдҪ е°ұжҳҜдёҖдёӘ专家гҖӮ& \) t* R5 a8 X5 h% w
- S' B( T# p+ b7 n
еҪ“дҪ еҸ‘еұ•еҲ°дәҶжӣҙй«ҳзҡ„еұӮж¬ЎпјҢдёҚд»…иғҪжӢ’з»қдёҚиҜҘеҒҡзҡ„жүӢжңҜпјҢиҝҳиғҪдёәз—…дәәжҸҗдҫӣжӣҙеҘҪзҡ„жІ»з–—зӯ–з•ҘпјҢжҲ‘и§үеҫ—иҝҷеҸҜд»Ҙз§°дёәеӨ§еёҲдәҶгҖӮ
8 y! ]6 k1 A8 Y) e ~% g5 g+ GжүҖд»ҘиҜҙеӨ–科еҢ»з”ҹжңүдёҚеҗҢзҡ„зә§еҲ«гҖӮ
, q! I0 z/ o: a. F9 L; d8 QеҚ•зәҜең°жҠҠжүӢжңҜеҒҡиө·жқҘпјҢеҸӘжҳҜжүӢжңҜеҢ зҡ„ж°ҙе№іпјӣ' z9 V3 p, I& {
еҪ“дҪ зҹҘйҒ“дёәд»Җд№ҲиҜҘеҺ»еҒҡпјҢдҪ е°ұеҸҜд»ҘеҪ“иҖҒеёҲдәҶпјӣ
" T E2 j0 ], M% @2 E- WеҪ“дҪ иғҪжҠҠзҺ°жңүзҡ„зҹҘиҜҶжңүжңәең°з»„еҗҲеҲ°ж–°зҡ„зӯ–з•ҘйҮҢпјҢдҪ е°ұжҳҜеӨ§еёҲзә§зҡ„дәәзү©гҖӮ
* @! l) Z7 ^" U( ~3 Y
; L/ ]$ o2 e4 N' c, m- r, _, K! ~( q
еӣ жӯӨпјҢжҢүз…§жҲ‘зҡ„зҗҶи§ЈпјҢдёҖдёӘзҺ°д»Јзҡ„иғёеӨ–科еҢ»з”ҹдёҚеә”иҜҘеҸӘеұҖйҷҗдәҺеӨ–科жҠҖжңҜжң¬иә«пјҢ
3 {% Z* `* p" r+ `зү№еҲ«жҳҜиӮҝзҳӨеӨ–科еҢ»з”ҹпјҢиҰҒдёҚж–ӯжӢ“е®ҪзҹҘиҜҶйқўпјҢдёҚж–ӯдәҶи§Је…¶д»–еӯҰ科зҡ„еҸ‘еұ•гҖӮ: q' R: M3 B" M/ {8 S; Y1 ]# _. {9 b
еҪ“然пјҢеҜ№дәҺе…·дҪ“жҹҗдёҖйўҶеҹҹзҡ„дәҶи§ЈдёҚйңҖиҰҒеҫҲиҜҰе°ҪпјҢдҪҶжҳҜеҝ…йЎ»иҰҒзҹҘйҒ“жҲ‘们зҡ„иҚҜзү©иғҪеҸ‘еұ•еҲ°д»Җд№ҲзЁӢеәҰпјҢ- H( o% Y# D* d" }. l# h
жҲ‘们зҡ„ж”ҫе°„жІ»з–—иғҪеҸ‘еұ•еҲ°д»Җд№ҲзЁӢеәҰпјҢжҲ‘们иҝҳиҰҒеҸ‘еұ•иӮҝзҳӨзҡ„з”ҹзү©еӯҰгҖӮ
) B. g: V' J3 Y2 X( A# D% J D& V, [2 o& e5 b$ N
иҝ‘еҮ е№ҙеӣҪйҷ…дёҠеҮәзҺ°дәҶдёҖдёӘж–°еҗҚиҜҚпјҢеҸ«еҒҡвҖңmolecular surgeonвҖқпјҢд№ҹе°ұжҳҜвҖңеҲҶеӯҗеӨ–科еҢ»з”ҹвҖқ
1 }6 G. F6 } d7 ~; hгҖӮеҲҶеӯҗеӨ–科еҢ»з”ҹжҳҜд»Җд№Ҳе‘ўпјҹжңүдёҖжү№еӨ–科еҢ»з”ҹпјҢ他们дёҚдҪҶжүӢжңҜеҒҡзҡ„йқһеёёзІҫж№ӣпјҢиҖҢдё”еҜ№з”ҹзү©еӯҰиЎҢдёәд№ҹдәҶи§Јзҡ„йқһеёёйҖҸеҪ»пјҢ
0 K( F4 @0 o+ A% YеңЁиҝҷдёӘжҢҮеҜјдёӢе°ұдә§з”ҹдәҶиҙЁзҡ„йЈһи·ғпјҢиҝҷеҜ№жІ»з–—зӯ–з•Ҙе°ұдә§з”ҹйқһеёёеӨ§зҡ„ж”№еҸҳгҖӮжүҖд»ҘиҜҙmolecular surgeonжҳҜдёҖдёӘйқһеёёеҘҪзҡ„иҜҚпјҢ3 Y- M) k2 I" l l ^* M f; ]
е®ғе‘ҠиҜүжҲ‘们еҢ»з”ҹиҰҒеҫҖе“ӘдёӘж–№еҗ‘еҺ»иө°пјҢеҫҖе“ӘдёӘж–№еҗ‘еҺ»еҸ‘еұ•гҖӮ% m' ^1 \" P7 y9 L# y
+ r2 C4 G1 f0 I$ s) D8 QеӨ–科еҢ»з”ҹзҡ„иҒҢдёҡз”ҹж¶ҜеҫҲеӨ§зЁӢеәҰдёҠжҳҜйқ жҠҖжңҜж”Ҝж’‘зҡ„гҖӮ$ `, u. n! j. g4 B
жҲ‘и®Өдёә第дёҖдёӘйҳ¶ж®өпјҢд№ҹе°ұжҳҜеӨҙеҚҒе№ҙпјҢеә”иҜҘд»ҘжҠҖжңҜеҹ№и®ӯдёәдё»гҖӮ
6 P& J- s H7 q5 [4 ?6 P3 i- W+ M! dеҪ“然пјҢжҠҖжңҜеҹ№и®ӯзҡ„еҗҢж—¶д№ҹиҰҒзҒҢиҫ“е…ЁеұҖзҡ„и§ӮеҝөпјҢдҪҶжҳҜдёҚдёҖе®ҡиҰҒиҫ№еҒҡжҠҖжңҜжҖ§ж“ҚдҪңиҫ№еҒҡе…¶д»–зҡ„иҪ¬еҢ–жҖ§з ”究пјҢ1 `2 a5 b! ~4 J# {' u! t9 X
еӣ дёәйӮЈжҳҜдёҚзҺ°е®һзҡ„пјҢеҹ№и®ӯеҸӘиғҪеҲҶйҳ¶ж®өиҝӣиЎҢгҖӮ
- C6 D$ K- g, H) X; ^& [& ]
! T! ?- r( L4 o; kдҪҶжҳҜеҰӮжһңдёҖдҪҚеҢ»еёҲзҡ„иҒҢз§°еҲ°дё»жІ»еҢ»еёҲд»ҘдёҠпјҢз”ҡиҮіеҲ°еүҜй«ҳпјҢ4 h+ v7 P F, {/ ^7 @& a1 x
д»–е°ұеҝ…йЎ»е…·еӨҮжӣҙеҠ е№ҝжіӣзҡ„зҹҘиҜҶйқўпјҢжҲ‘们еҜ№д»–зҡ„еҹ№е…»дёҚд»…жҳҜжҠҖжңҜжң¬иә«пјҢиҖҢдё”иҰҒеҹ№е…»е…ЁеұҖзҡ„и§ӮеҝөпјҢ `/ O' G- L) u& O
еҜ№scienceпјҲ科еӯҰпјүзҡ„иҝҪеҜ»пјҢеҜ№й—®йўҳзҡ„жҺўзҙўпјҢеҜ№еҲҶеӯҗз”ҹзү©еӯҰжҠҖжңҜзҡ„жҺҢжҸЎпјҢиҝҷжҳҜ第дәҢдёӘйҳ¶ж®өгҖӮ% g# m8 ?, j* o5 @1 [
; A% g5 I4 R$ d然еҗҺпјҢеҜ№дәҺдёҖдҪҚдё»д»»зҡ„еҹ№е…»пјҢеә”иҜҘжіЁйҮҚеҜ№ж•ҙдёӘи§ҶйҮҺзҡ„еҹ№и®ӯпјҢиҝҷжҳҜдёҚж–ӯеӯҰд№ зҡ„иҝҮзЁӢгҖӮ
' G# n# p* p8 c$ E7 I9 DеңЁж•ҙдёӘиҝҮзЁӢдёӯж°ёиҝңдёҚиғҪеҝҪз•Ҙзҡ„е°ұжҳҜдәәж–ҮзІҫзҘһпјҢдёҚз®Ўд»ҺеӯҰж ЎеҲ°иө°иҝӣзӨҫдјҡпјҢд»ҺдҪ ејҖе§ӢеҪ“дёҖдёӘеӨ–科еҢ»з”ҹпјҢе°ұеҝ…йЎ»ж°ёиҝңжңүдәәж–ҮзІҫзҘһгҖӮ
% V& j* _) B4 J' G- g
9 k' L# n1 g- k! O5 h4 R1 GдёҖдёӘеӨ–科еҢ»з”ҹзҡ„дјҰзҗҶеә•зәҝжҳҜд»Җд№ҲпјҹеҜ№з—…дәәжңүеҲ©зҡ„жҲ‘们жүҚеҺ»еҒҡпјҢеҜ№з—…дәәдёҚеҲ©зҡ„жҲ‘们дёҚиғҪеҺ»еҒҡпјҢ$ [ c; O) L- S/ b" b8 ?6 v# g
дёҚз®ЎдҪ еҜ№иҝҷдёӘжҠҖжңҜеӨҡж„ҹе…ҙи¶ЈгҖӮеӣ жӯӨпјҢжҲ‘们зҡ„еҹ№е…»жңүдёӘиҝҮзЁӢпјҢжңүзҡ„жҳҜз»Ҳиә«еҹ№е…»дёӢеҺ»пјҢжңүзҡ„жҳҜеҲҶйҳ¶ж®өиҝӣиЎҢзҡ„гҖӮ
4 l3 R- i1 O! V0 O6 p. q) ]; F; @5 Y, d8 S' l: j
дёҒйҰҷеӣӯпјҡжңҖеҗҺиҜ·и°Ҳи°ҲжӮЁеҜ№иҝҷж¬ЎеӨ§дјҡзҡ„ж„ҹеҸ—гҖӮ
; s$ C2 ~, C* J- [6 X
/ K6 P' M! o! o7 P" ]еҗҙдёҖйҫҷж•ҷжҺҲпјҡжң¬ж¬Ўдјҡи®®йҮҚеңЁз»јеҗҲжІ»з–—пјҢеҪ“然жҳҜд»ҘеӨ–科дёәдё»пјҢиҝҷйқһеёёз¬ҰеҗҲжҲ‘иҮӘе·ұзҡ„зҗҶеҝөгҖӮ" d9 ^% X: Z6 U- s3 u
дёҖдёӘеӨ–科еҢ»з”ҹеңЁжҲҗй•ҝзҡ„иҝҮзЁӢдёӯиҰҒдёҚж–ӯең°зҒҢиҫ“з»јеҗҲжІ»з–—зҡ„зҗҶеҝөпјҢдёҚж–ӯең°зҒҢиҫ“еҜ№жңҖж–°зҹҘиҜҶзҡ„жёҙжұӮпјҢеҜ№жңҖж–°зҹҘиҜҶзҡ„жҺҢжҸЎпјҢиҝҷжҳҜеӨ§дјҡжҲҗеҠҹзҡ„ең°ж–№гҖӮ1 Q# g- t# z5 f: a$ R
! T9 n; L6 Z" o Z
дёҒйҰҷеӣӯпјҡйқһеёёж„ҹи°ўеҗҙж•ҷжҺҲжҺҘеҸ—жҲ‘们зҡ„йҮҮи®ҝпјҒ |

 [еӨҚеҲ¶й“ҫжҺҘ]
[еӨҚеҲ¶й“ҫжҺҘ]


 еҗҢжӯҘж”ҫеҢ–з–—з»“жқҹ3е№ҙпјҢеҢ–е…ҚиҒ”еҗҲеҗҺеҚ•е…Қ
жҒіиҜ·еҗ„дҪҚеё®еҝҷжҢҮеҜјпјҒ
2021е№ҙ7жңҲејҖе§Ӣж №жІ»жҖ§и°ғејәж”ҫз–—пјҢжҖ»и®Ў33ж¬ЎгҖӮиҝ‘4е№ҙжІ»з–—ж–№жЎҲиҫғз®ҖеҚ•пјҢ
еҗҢжӯҘж”ҫеҢ–з–—з»“жқҹ3е№ҙпјҢеҢ–е…ҚиҒ”еҗҲеҗҺеҚ•е…Қ
жҒіиҜ·еҗ„дҪҚеё®еҝҷжҢҮеҜјпјҒ
2021е№ҙ7жңҲејҖе§Ӣж №жІ»жҖ§и°ғејәж”ҫз–—пјҢжҖ»и®Ў33ж¬ЎгҖӮиҝ‘4е№ҙжІ»з–—ж–№жЎҲиҫғз®ҖеҚ•пјҢ
 иӮәйіһзҷҢL858R иҫҫе…Ӣжӣҝе°јиҝ‘дёүе№ҙиҖҗиҚҜ
71еІҒпјҢеҘіпјҢж— еҗёзғҹеҸІ
18е№ҙ10жңҲиӮәйіһзҷҢпјҢ3X 4cmпјҢе·ҰиӮәдёҠеҸ¶еҲҮйҷӨпјҢж— иҪ¬з§»пјҢжңӘеҢ–з–—
22е№ҙ1жңҲ
иӮәйіһзҷҢL858R иҫҫе…Ӣжӣҝе°јиҝ‘дёүе№ҙиҖҗиҚҜ
71еІҒпјҢеҘіпјҢж— еҗёзғҹеҸІ
18е№ҙ10жңҲиӮәйіһзҷҢпјҢ3X 4cmпјҢе·ҰиӮәдёҠеҸ¶еҲҮйҷӨпјҢж— иҪ¬з§»пјҢжңӘеҢ–з–—
22е№ҙ1жңҲ
 еҘҘиҘҝжӣҝе°јиҖҗиҚҜеҗҺз»ӯжІ»з–—пјҢеҮәзҺ°и…№ж°ҙ
жІ»з–—з»ҸиҝҮпјҡ
2010зЎ®иҜҠи…әзҷҢ
жІ»з–—ж–№жЎҲпјҡеҸіиӮәдёҠеҸ¶еҲҮйҷӨпјҢ10е№ҙж°‘иҜәе®ҫ2ж¬ЎпјҢйЎәй“Ӯ+зӣ–иҜә2ж¬ЎгҖӮ
еҘҘиҘҝжӣҝе°јиҖҗиҚҜеҗҺз»ӯжІ»з–—пјҢеҮәзҺ°и…№ж°ҙ
жІ»з–—з»ҸиҝҮпјҡ
2010зЎ®иҜҠи…әзҷҢ
жІ»з–—ж–№жЎҲпјҡеҸіиӮәдёҠеҸ¶еҲҮйҷӨпјҢ10е№ҙж°‘иҜәе®ҫ2ж¬ЎпјҢйЎәй“Ӯ+зӣ–иҜә2ж¬ЎгҖӮ
 дёүд»ЈдјҸзҫҺжҚўдёҖд»Јзү№зҪ—еҮҜиҺ·зӣҠзҺҮй—®йўҳ
家зҲ¶25е№ҙ2жңҲеӣ иӮ©иҶҖз–јз—ӣзЎ®иҜҠиӮәи…әзҷҢжҷҡжңҹ4BпјҢеҸҢиӮәиҪ¬пјҢеӨҡеҸ‘йӘЁиҪ¬пјҢи„‘иҪ¬пјҲйқһе…ёеһӢпјүпјҢ
дёүд»ЈдјҸзҫҺжҚўдёҖд»Јзү№зҪ—еҮҜиҺ·зӣҠзҺҮй—®йўҳ
家зҲ¶25е№ҙ2жңҲеӣ иӮ©иҶҖз–јз—ӣзЎ®иҜҠиӮәи…әзҷҢжҷҡжңҹ4BпјҢеҸҢиӮәиҪ¬пјҢеӨҡеҸ‘йӘЁиҪ¬пјҢи„‘иҪ¬пјҲйқһе…ёеһӢпјүпјҢ
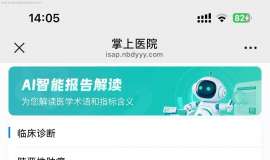
 зӣІиҜ•йқ¶еҗ‘иҚҜ 29дёӘжңҲпјҢжІ»з–—еҲҶдә«
ж—¶й—ҙпјҡ2025/1/27 зӣІиҜ•йқ¶еҗ‘иҚҜ第29дёӘжңҲгҖӮ
жң¬жңҲжІ»з–—ж–№жЎҲпјҡ7080пјҲ14mgпјүгҖӮ2024е№ҙ11жңҲеә•
зӣІиҜ•йқ¶еҗ‘иҚҜ 29дёӘжңҲпјҢжІ»з–—еҲҶдә«
ж—¶й—ҙпјҡ2025/1/27 зӣІиҜ•йқ¶еҗ‘иҚҜ第29дёӘжңҲгҖӮ
жң¬жңҲжІ»з–—ж–№жЎҲпјҡ7080пјҲ14mgпјүгҖӮ2024е№ҙ11жңҲеә•



 жҳҫиә«еҚЎ
жҳҫиә«еҚЎ